Transcriptomics Sequencing Services
 Inquire
InquireCatalog Number|Packaging
Mat. No |
Ref. No |
No. of preps |
-
Description
TIANGEN transcriptome sequencing can comprehensively and quickly obtain almost all transcribed sequence information of a specific tissue or organ in a certain state. In bioinformatic analysis, comparison with the reference genome leads to the comprehensive and quick obtaining of mRNA sequence and expression abundance information, as well as to analysis of structures such as fusion genes and alternative splicing.
-
Technical Workflow
Scheme design → Sample processing → Library construction and sequencing → Data analysis → After-sales service
-
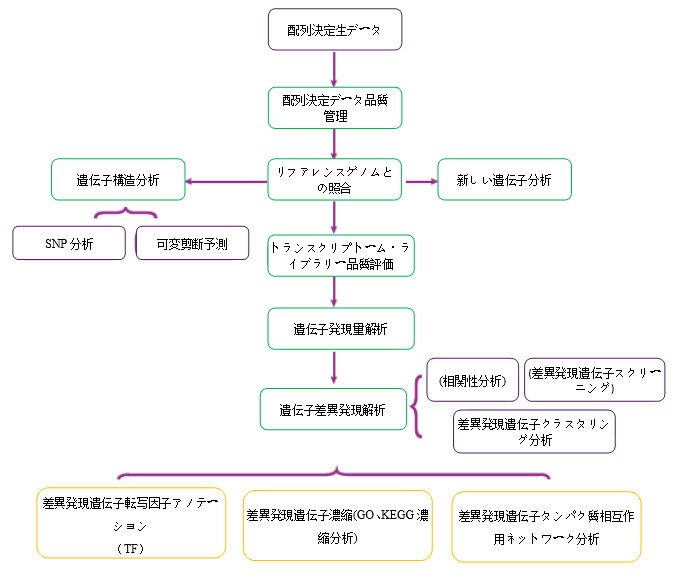
Analysis Workflow
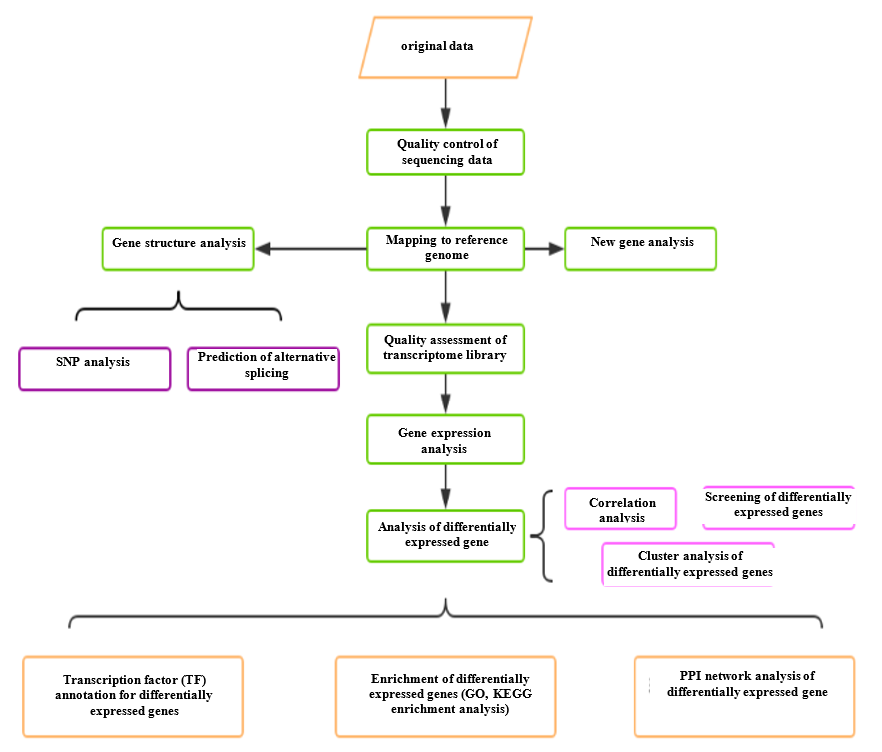
-
Features
1. As a global supplier of high-quality NGS raw materials, we guarantee the quality of the project from the source, including high-quality extraction and library construction etc.;
2. Senior technical teams, with more than ten years of information analysis experience, can provide a variety of personalized analysis;
3. Powerful database support, with databases such as TCGA and HGMD;
4. With high-quality after-sales service, no longer need to worry about the results anymore;
-
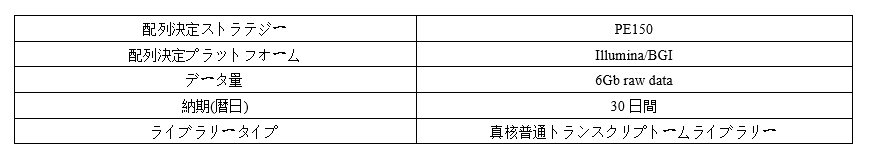
Parameter

-
Applications
Environmental threats, agronomic traits, medicine development, molecular markers development, growth and development, etc.
-
Requirements of Samples
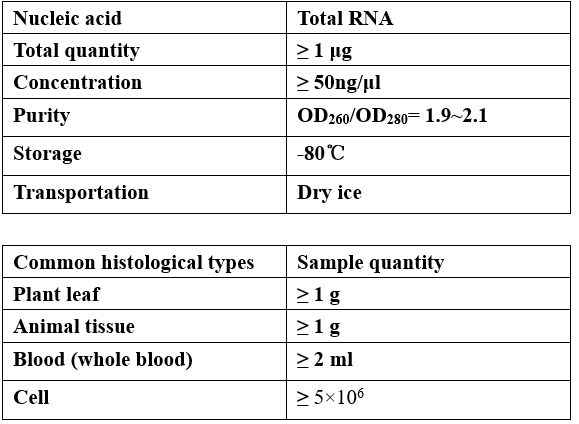
-
Results Demo
Differentially expressed volcano plot
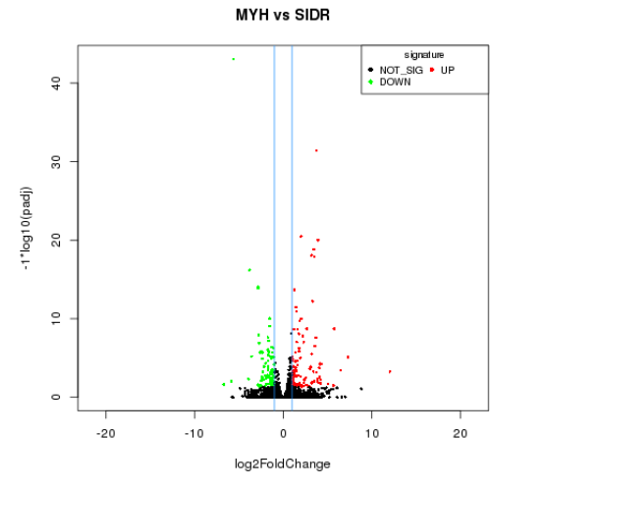
The input data of differentially expressed gene is the readcount data obtained in the analysis of gene expression level. For samples with biological replicates, we use deseq for analysis; For samples without biological replicates, we use edgeR for analysis. Finally, we select genes with FDR (false discovery rate) less than 0.05 and fold change (FC) greater than or equal to 2 as the differentially expressed genes.
Cluster heat map

Cluster analysis is used to judge the expression patterns of differential genes under different experimental conditions; By grouping genes with the same or similar expression patterns into clusters, the functions of unknown genes or unknown functions of known genes can be identified; Because these similar genes may have similar functions or participate in the same metabolic process or cellular pathway.
GO enrichment
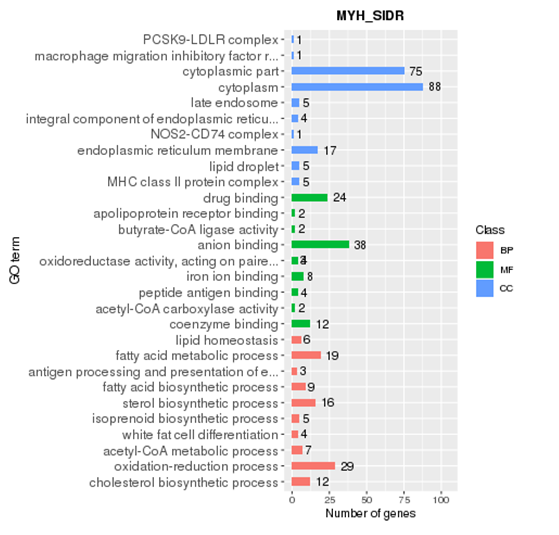
Gene Ontology (referred to as GO) is an international standard classification system for gene function. After screening the differential genes according to the experimental purpose, the distribution status of the differential genes in GO was studied in order to clarify the expression of the sample differences in the gene function in the experiment. The GO annotation information of most species is downloaded from Ensembl's Biomart database (http://asia.ensembl.org). If there is no GO annotation information for this species on Ensembl, the GO information annotated by yourself will be used.
PPI network analysis

Use the interaction relationship in the STRING PPI database (http://string-db.org/) to directly extract the interaction relationship of the target gene set (such as the differential gene list) from the database for the species contained in the database to build a network; For species not included in the database, first, we use Blastx to compare the target gene set sequence to the protein sequence of the closely related or model species contained in the STRING database, and construct the interaction network by using the PPI relationships of the selected related or model species.

-
Sort by
-
Date
 Date()
Date()
Date
 Date()
Date()
Impact Factor
IF()
Impact Factor
IF()